Flex Funds Project: nf-workflows
Reproducible workflows with nextflow and nf-core (nf-workflows)
Project Start and End Date
2025-01-01 - 2025-12-31
Short project summary
The project focused on the maintenance and targeted extension of selected nf-core data analysis workflows relevant to the NFDI4Microbiota community, complemented by training and user support activities. nf-core is a community-driven collection of standardized, peer-reviewed bioinformatics workflows built using Nextflow, a workflow management system that enables scalable, portable, and reproducible data analysis across diverse computing environments. Together, Nextflow and nf-core provide a widely adopted framework for implementing robust and FAIR-aligned bioinformatics pipelines.
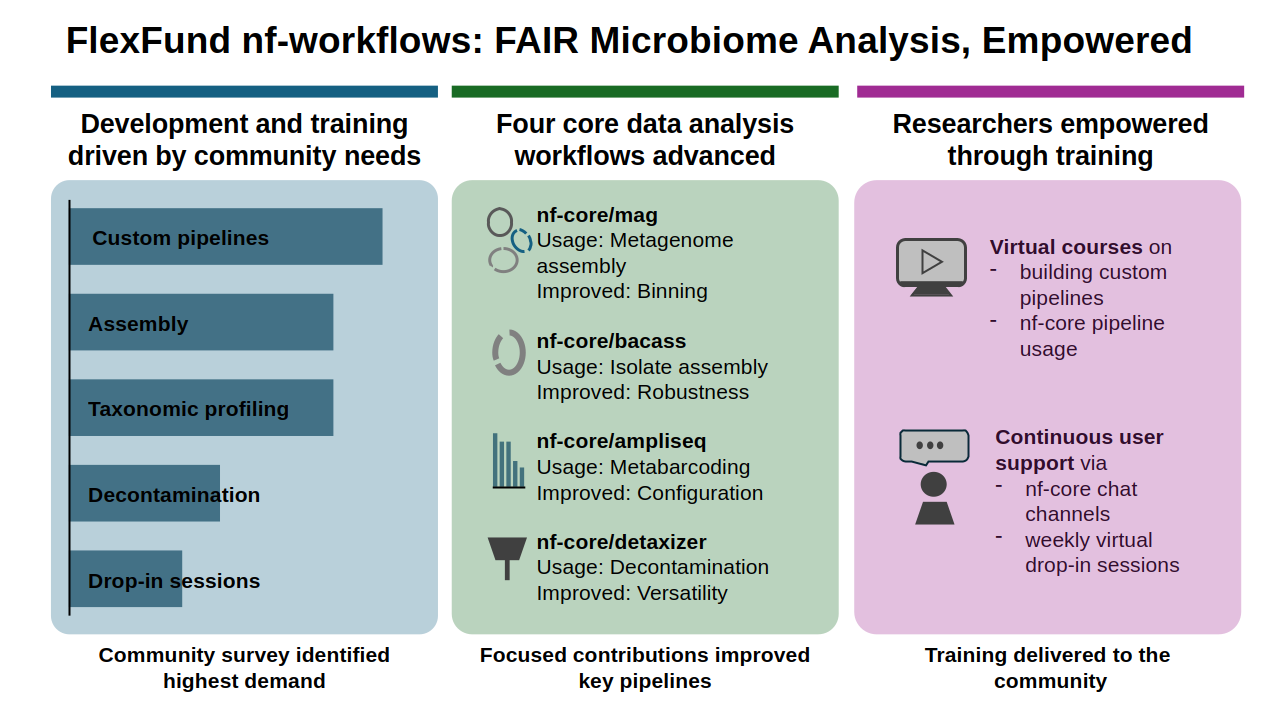
To align development with community needs, a survey was conducted in collaboration with Sina Barysch (EMBL Heidelberg; de.NBI e.V.), identifying strong demand for workflows addressing metagenomic assembly, taxonomic profiling, and data decontamination. Based on these results, focused contributions were made to four nf-core workflows. The shotgun metagenomics workflow nf-core/mag was extended with additional binning algorithms informed by CAMI II benchmarking and expert input from Alice McHardy’s group (HZI). Continuous integration tests were added to the small-genome assembly workflow nf-core/bacass to improve robustness and long-term maintainability. The nf-core/ampliseq workflow for taxonomic profiling underwent extensive maintenance, performance optimization, and updates to default reference databases. The workflow nf-core/detaxizer, used for removing contaminant sequences from metagenomic data, was substantially improved and documented in a peer-reviewed publication.
Beyond workflow development, the project placed strong emphasis on training, support, and accessibility. Based on surveyed needs, a virtual course on building custom Nextflow-based pipelines was delivered, complemented by general nf-core training at community events. Continuous user support was provided via online chat and weekly drop-in sessions and promoted through multiple NFDI4Microbiota communication channels. Overall, the project lowered barriers to reproducible bioinformatics analyses, empowered non-specialist users, and delivered immediately usable, FAIR-aligned workflows that directly benefit the NFDI4Microbiota community.
Graphical abstract
Brief summary of the main results and conclusion
The project significantly improved several key nf-core workflows used by the NFDI4Microbiota community, including metagenomic assembly, taxonomic profiling, and data decontamination pipelines. Community-driven prioritization ensured that development addressed real user needs, while benchmarking-based improvements and extensive maintenance increased robustness, performance, and sustainability. In parallel, targeted training and continuous support empowered researchers to build, adapt, and confidently use Nextflow- and nf-core–based workflows. Overall, the project strengthened reproducible microbiome data analysis, lowered technical entry barriers, and provided durable benefits to both the NFDI4Microbiota community and the wider bioinformatics ecosystem.
<< Back to all past Flex FundsProject Members

Dr. Daniel Straub
ORCID ID: 0000-0002-2553-0660
University of Tübingen
Dr. Sven Nahnsen
ORCID ID: 0000-0002-4375-0691
University of Tübingen
Dr. Jannik Seidel
ORCID ID: 0009-0003-2867-2335
University of Tübingen
Keywords
workflows
nextflow
nf-core
community
bioinformatics
FAIR